

Livedovaskulopathie
Die Livedovaskulopathie ist eine chronisch rezidivierende Gefäßerkrankung der unteren Extremitäten. Sie entsteht durch Thrombosen in der Mikrozirkulation, was zu einer verminderten Durchblutung führt.
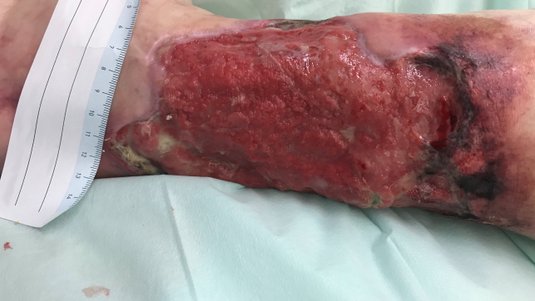
In Folge kommt es zu der typischen Hautveränderung, dem netzartigen, marmorierten Muster der Livedo racemosa. Im fortgeschrittenen Stadium können zudem schmerzhafte Hautulzerationen auftreten.
Die Livedovaskulopathie ist ein komplexes Krankheitsbild, das durch eine gestörte Blutzirkulation in den Hautgefäßen verursacht wird. Diese führt zu der typischen Hautveränderung an den unteren Extremitäten, der Livedo racemosa. Im fortgeschrittenen Krankheitsstadium können zusätzlich schmerzhafte Ulzerationen auftreten. In der medizinischen Literatur gibt es eine Vielzahl von Begriffen, die dieses Krankheitsbild beschreiben. Zu den häufig verwendeten Begriffen gehören Livedo reticularis mit Sommerulzerationen, hyalinisierende Vaskulitis, PPURPLE (painful purpuric ulcers with reticular pattern of lower extremities), Livedovaskulitis sowie idiopathische „Atrophie blanche“.
Wie wird die Livedovaskulopathie behandelt?
Wundtherapie
Ulzerationen sind Substanzdefekte der Haut oder Schleimhaut. Bei der Haut reicht die Schädigung bis in die Dermis hinein oder noch tiefer. Ulzera zeichnen sich häufig durch eine langwierige Heilungsphase aus und bedürfen einer individuell abgestimmten Behandlung aus geeigneter Wundreinigung und modernen Wundauflagen. Die Leitlinie „Diagnostik und Therapie der Livedovaskulopathie“ empfiehlt zudem im seltenen Fall einer bakteriellen Superinfektion eine zusätzliche antibiotische Therapie. Supportive Maßnahmen bei der Behandlung der Livedovaskulopathie umfassen eine Kompressionstherapie, falls von der Patientin oder dem Patienten toleriert, sowie den Verzicht auf große Temperaturschwankungen, um Stase (Rückstau) zu vermeiden. Für therapierefraktäre Ulzerationen kann der Einsatz von einem gewebespezifischen Plasminogenaktivator zur Aktivierung der Fibrinolyse erwogen werden. In Einzelfällen kann eine hyperbare Sauerstofftherapie zur Schmerzreduktion und Abheilung aktiver Läsionen eingesetzt werden. Allerdings erzielt diese Maßnahme lediglich eine vorübergehende Verbesserung der Symptomatik. Nach dem Ende der Behandlung können Rezidive auftreten, meist etwa 6 Monate später.
Medikamentöse Therapie
Um das Fortschreiten der chronischen Minderdurchblutung mit Gewebsischämie und Narbenbildung zu vermeiden, ist der frühzeitige Beginn einer medikamentösen Therapie notwendig. Die Leitlinie „Diagnostik und Therapie der Livedovaskulopathie“ empfiehlt in der Erstlinientherapie eine Antikoagulation („Blutverdünner“) mit niedermolekularem Heparin oder Rivaroxaban/DOAK zur Verhinderung der Thrombosen. Ebenso können intravenöse Immunglobuline (IVIG) verabreicht werden. Beide Behandlungen erfolgen off-label.
Bei Therapierefraktärität oder Vorliegen von assoziierten Erkrankungen empfiehlt die Leitlinien den Einsatz von Phenprocoumon/Warfarin (Vitamin-K-Antagonisten), Iloprost, Vitamin B6, B12 und Folsäure in der Zweitlinie. Bei assoziierten Autoimmunerkrankungen sollte der Einsatz von Glukokortikoiden zur Therapie dieser Autoimmunerkrankung erfolgen. Im Hinblick auf eine supportive Schmerztherapie kann ebenfalls der Einsatz von nichtsteroidalen Antirheumatika (NSAR) erwogen werden.
Der Begriff «Livedo» beschreibt eine erythematös-livide Hautverfärbung, die durch eine Beeinträchtigung des Blutflusses in den Hautgefäßen entsteht. Es lassen sich zwei Formen unterscheiden: die meist harmlose Livedo reticularis und die pathologische Livedo racemosa. Die Livedo reticularis tritt vor allem bei jungen Frauen als reversible Erscheinung auf. Sie betrifft meist die unteren Extremitäten, geht mit lividen, geschlossen-netzartigen Hautzeichnungen einher und ist stress- und/oder temperaturabhängig. Im Gegensatz zur Livedo reticularis ist die Livedo racemosa ein seltenes, persistierendes Symptom, das auf eine zugrunde liegende strukturelle oder systemische Erkrankung hinweist. Die typischen diskontinuierlichen, blitzfigurenartigen Hautzeichnungen der Livedo racemosa können den Manifestationen der Grunderkrankung Monate oder sogar Jahre vorausgehen.
Was sind Symptome bei Livedovaskulopathie?
Die Livedovaskulopathie verläuft in drei typischen, rezidivierenden Stadien. Im ersten Stadium zeigt sich eine Livedo racemosa, eine netzförmige, livide Zeichnung, die durch eine Minderdurchblutung der Dermis entsteht.
Im zweiten Stadium führt die Minderdurchblutung zu Gewebsischämie und Ulzerationen. Den Ulzerationen geht typischerweise ein brennender, punktueller Schmerz voraus, der als „Angina cutis“ bezeichnet wird. Der Schmerz tritt üblicherweise 1-3 Tage vor den Nekrosen auf, die mit hämorrhagischen (blutungsbedingten) Blasen und Ulzerationen verbunden sind. Wird in dem Zeitfenster zwischen der Angina cutis und dem Auftreten von Nekrosen eine schnelle Therapie eingeleitet, kann eine Gewebenekrose häufig noch verhindert werden. Bei einem Überschreiten der Phase ohne Behandlung manifestieren sich die Nekrosen und Blasen. Im weiteren Verlauf entstehen Ulzerationen mit Krustenbildung.
Im dritten Stadium stellt die „Atrophie blanche“ die chronische Manifestation und den Endpunkt narbiger Umbauprozesse dar. Sie erscheint als blitz- oder sternförmige, porzellanfarbene Narbe.
Die Klinik der Livedovaskulopathie ist wesentlich durch die beschriebene Trias aus Livedo racemosa, Ulzerationen und „Atrophie blanche“ gekennzeichnet, wobei diese Symptome auch isoliert oder kombiniert auftreten können. Der Verlauf ist chronisch, mit Phasen der Abheilung und neuen Schüben. Eine neurologische Beteiligung mit Dys- und Hypoästhesien (Empfindungsstörungen) ist möglich. Die Livedovaskulopathie tritt zwar ganzjährig auf, jedoch können saisonale Schwankungen in der Symptomatik beobachtet werden.
Diagnose der Livedovaskulopathie
Die Diagnose wird klinisch und histologisch gestellt. Für die Feststellung der zugrundeliegenden Pathogenese wird eine Gerinnungsdiagnostik mit Screening nach prokoagulatorischen (gerinnungsfördernden) Parametern empfohlen.
Folgende klinische Aspekte sollten für die Diagnosestellung der Livedovaskulopathie berücksichtigt werden: Charakteristisch ist die Trias aus Livedo racemosa, Ulzerationen und „Atrophie blanche“. Die Erkrankung tritt ganzjährig auf und betrifft Frauen häufiger als Männer. Typischerweise wird das Auftreten neuer Ulzerationen von einem brennenden Schmerz in der betroffenen Region eingeleitet, der sich auf den Manifestationsort beschränkt.
Die histologische Diagnose der Livedovaskulopathie ist nur im Akutstadium möglich. Idealerweise wird dazu eine Spindelbiopsie aus dem Randbereich des betroffenen Areals entnommen. Typisch sind Fibrinablagerungen in den Gefäßwänden und Fibrinthromben in Gefäßen der oberen und mittleren Dermis. Prominente Entzündungen oder Gefäßwandnekrosen deuten eher auf eine Vaskulitis (Gefäßentzündung) hin. Das Stadium der „Atrophie blanche“ zeichnet sich durch eine gefäßarme Narbe mit einer dünneren, atrophierten Epidermis aus. Zusätzlich kann eine Umstrukturierung der Thromben auftreten.
Differentialdiagnose
Die Differenzialdiagnose der Livedovaskulopathie erfolgt durch ihre typische Klinik (stechende Schmerzen, ganzjähriges Auftreten) und die charakteristische Histologie im frühen Stadium.
Wichtige Differenzialdiagnosen umfassen:
Periphere arterielle Verschlusskrankheit (pAVK) mit einhergehendem Ulcus cruris arteriosum: Patienten und Patientinnen mit pAVK zeigen ebenfalls häufig an den Beinen gleichzeitig Livedo racemosa-artige Erytheme, Ulzera und eine „Atrophie blanche“. Durch eine angiologische Untersuchung kann die pAVK von der Livedovaskulopathie unterschieden werden.
Ulcus cruris venosum: Das Ulcus cruris venosum entwickelt sich langsamer, zeigt gelblichbraune Hyperpigmentierungen und sichtbare Varikosis sowie eine Corona phlebectatica paraplantaris.
Kutane Polyarteriitis nodosa (PAN): Die Kutane Polyarteriitis nodosa ist eine Entzündung der mittelgroßen Arterien. Die Erkrankung geht ebenfalls mit einer Livedo racemosa einher, jedoch treten tastbare subkutane Nodi auf.
Pyoderma gangraenosum: Das Pyoderma gangraenosum ist eine chronisch entzündliche Hauterkrankung unbekannten Ursprungs. Sie unterscheidet sich von der Livedovaskulopathie durch den lividen Randsaum, eine schnelle Größenzunahme und gutes Ansprechen auf Glukokortikoide oder Ciclosporin.
Weitere Differenzialdiagnosen umfassen Sneddon-Syndrom, Antiphospholipid-Syndrom, Kalziphylaxie, ANCA-positive Vaskulitiden, Kryoglobulinämie und medikamentös verursachte Ulzerationen.

Grunderkrankungen bei Livedo racemosa
Livedo racemosa kann in Verbindung mit verschiedenen Erkrankungen auftreten, unter anderem:
Antiphospholipid-Syndrom
Das Antiphospholipid-Syndrom ist eine systemische Autoimmunerkrankung, die mit Thrombosen und Entzündungsreaktionen einhergeht. Ausgelöst wird die Erkrankung durch eine heterogene Gruppe von erworbenen Antikörpern. Klinisch manifestiert sich das Antiphospholipid-Syndrom meist durch venöse und arterielle Thrombosen sowie Schwangerschaftskomplikationen. Zusätzlich können pathologische Manifestationen wie neurologische Dysfunktion, dermatologische Symptome wie Livedo racemosa, Thrombozytopenie, Herzklappenerkrankungen sowie Nierenerkrankungen auftreten.
Kalciphylaxie
Bei der Kalziphylaxie kommt es zu einer vaskulären Kalzifikation, die typischerweise durch den Verschluss von Mikrogefäßen im Hautgewebe gekennzeichnet ist und zu schmerzhaften Hautläsionen führt. Die Erkrankung betrifft in der Regel Patienten und Patientinnen mit Nierenerkrankungen im Endstadium. Zu den anfänglichen Symptomen gehören unter anderem Hautveränderungen wie Livedo racemosa, Plaques und Knötchen. Die Hautläsionen können sich schnell zu Ulzera entwickeln.
Sneddon-Syndrom
Das Sneddon-Syndrom ist eine seltene nicht-entzündliche thrombotische Vaskulopathie, die durch die Kombination aus einer zerebrovaskulären Krankheit und einer Livedo racemosa gekennzeichnet ist. Im Verlauf der Erkrankung sind Gedächtnisstörungen bis hin zur Demenz häufig. Das mittlere Erkrankungsalter beträgt für die neurologischen Symptome 39 Jahre. Die Livedo racemosa tritt meist 10 Jahre früher, gelegentlich auch schon in der Kindheit auf.
Systemischer Lupus erythematodes
Systemischer Lupus erythematodes ist eine chronische Autoimmunerkrankung aus der Gruppe der Kollagenosen. Livedo racemosa kann unspezifisch bei Patienten und Patientinnen mit Systemischen Lupus erythematodes auftreten, insbesondere in Zusammenhang mit vaskulären Entzündungen.
Es besteht auch die Möglichkeit, dass mehrere, mit Livedo racemosa assoziierte Erkrankungen komorbid auftreten. In vielen Fällen lässt sich allerdings keine zugrundeliegende Erkrankung diagnostizieren (idiopathische Livedo racemosa).
Epidemiologie – Wie häufig tritt die Livedovaskulopathie auf?
Die Livedovaskulopathie tritt häufiger bei Frauen als bei Männern auf, mit einem geschätzten Geschlechterverhältnis von 3:1. Die Inzidenz wird auf etwa 1:100.000 Personen pro Jahr geschätzt. Das Alter bei Erstdiagnose liegt üblicherweise zwischen 32 und 53 Jahren, wobei auch Fälle bei jungen Patientinnen bekannt sind. Als Kofaktoren gelten ein erhöhter Body-Mass-Index und arterielle Hypertonie, jedoch wurden bisher keine spezifischen Komorbiditäten identifiziert.
Pathogenese – Wie entsteht die Livedovaskulopathie?
Die Pathomechanismen der Livedovaskulopathie sind noch nicht vollständig geklärt. Jedoch ist bekannt, dass ein Ungleichgewicht in der Hämostase (Blutgerinnung) zugunsten prokoagulatorischer (gerinnungsfördernder) Faktoren zu Thrombusbildung in der Mikrozirkulation der Dermis führt. Dies verursacht kutane Ischämie (Minderdurchblutung), Gewebeinfarkt und Ulzerationen, während eingeschränkte Wundheilung und eingeschränkte Leukozytenmigration Wundinfektionen begünstigen.
Bei Patientinnen und Patienten mit Livedovaskulopathie wurden verschiedene prokoagulatorische Parameter wie erhöhte Antikardiolipinantikörper, Lupus-Antikoagulans sowie Mängel an Antithrombin III, Protein C und S identifiziert. Auch genetische Mutationen (z. B. Faktor V Leiden) und Stoffwechselstörungen wie erhöhte Lipoprotein(a)-Spiegel oder eine Beeinträchtigung des Homozysteinstoffwechsels steigern die Thromboseneigung. Dennoch lassen sich in weniger als der Hälfte der Fälle pathologische Veränderungen bekannter Gerinnungsfaktoren nachweisen, und umgekehrt führt nicht jede Anomalie zu Livedovaskulopathie.
Der Manifestationsort spielt ebenfalls eine Rolle, da ein reduzierter Perfusionsdruck und eine geringere Konzentration thrombolytischer Faktoren an der unteren Extremität das Thromboserisiko erhöhen.
Literatur

Nach ihrem Studium der Biologie an der Ruhr-Universität Bochum promovierte Dr. Lorenz zum Dr. rer. nat. Seit 2012 ist sie in der medizinisch-wissenschaftlichen Abteilung bei Dr. Ausbüttel tätig, seit 2018 auch als Leiterin dieser Abteilung sowie der Forschungsabteilung.
